Our Company
We are a clinical-stage biopharmaceutical company focused on creating value through (i) the streamlined development under Section 505(b)(2) of the Federal Food, Drug and Cosmetic Act, or FDCA of our lead product candidate, MAT9001, a highly purified, prescription-only omega-3 free fatty acid formulation specifically designed for the treatment of cardiovascular and metabolic conditions and (ii) the application of our lipid nano-crystal (LNC) platform delivery technology to solve complex challenges relating to the delivery of small molecules, gene therapies, vaccines, proteins and peptides. In general, the development timeline for a 505(b)(2) New Drug Application, or NDA, is shorter and less expensive than an NDA developed under Section 505(b)(1) for new chemical entities that have never been approved in the United States. Based upon MAT9001’s unique mixture of highly purified omega-3 free fatty acids and our observations of MAT9001’s enhanced bioavailability and potency as compared to Amarin Corporation’s Vascepa® (icosapent ethyl) in our initial head-to-head pharmacokinetic (PK) and pharmacodynamic (PD), or PK/PD, clinical study, we believe that the results of our forthcoming targeted clinical development activities and related clinical investigations may yield an improved therapeutic profile compared to currently-existing therapies.
MAT9001 is a soft gelatin capsule containing a complex mixture of polyunsaturated free fatty acids, including multiple long-chain omega-3 fatty acids, including primarily eicosapentanoic acid (EPA) and docosapentanoic acid (DPA). Amongst other properties, omega-3 fatty acids, which are also found in FDA-approved drugs in varying amounts, such as Vascepa®, have extensive clinical evidence of safety and efficacy in lowering triglycerides (TG) in patients with hypertriglyceridemia (HTG). We believe that based upon MAT9001’s unique composition, which includes more DPA than other known omega-3 fatty acids, it will prove to be differentiated from other existing therapies for the treatment of very high triglycerides, or severe hypertriglyceridemia (SHTG), and dyslipidemia.
Triglycerides are fats that are carried in the blood, together with cholesterol, within lipoproteins. High levels of triglyceride-rich lipoproteins are associated with an increased risk of atherosclerotic cardiovascular disease and in the case of severe hypertriglyceridemia, acute pancreatitis. High levels of triglycerides are due to both genetic and environmental factors and are associated with comorbid conditions such as diabetes, chronic renal failure, and nephrotic syndrome. Unlike the currently approved products in this category, many of which have been repurposed following clinical failures in their originally intended indications, we have specifically designed and developed MAT9001 to treat SHTG, dyslipidemia and other cardiovascular and metabolic conditions.
In 2015, we announced results from our head-to-head PK/PD clinical study against Vascepa, a prescription-only ethyl ester formulation of EPA. This was an open-label, cross-over design study conducted in 42 patients with elevated triglyceride levels. Patients were treated for 14 days with MAT9001 or Vascepa, (4 grams/day for both treatment arms), followed by a five-week wash-out period, then crossed over to the other treatment. The main objectives of the study were to measure the relative bioavailability of MAT9001 versus Vascepa as well as effects on triglyceride levels. In this study, we observed statistical superiority of MAT90001 in reducing serum triglycerides, total- and non-HDL-cholesterol, apolipoprotein CIII and PCSK9 levels. MAT9001 was observed to significantly reduce PCSK9 in patients. In this trial, MAT9001 achieved greater median percentage reduction in four of six lipid measures, including total cholesterol, when compared to Vascepa.
We are focusing our initial efforts on developing MAT9001 with an initial indication for the treatment of severe hypertriglyceridemia. If we receive U.S. Food and Drug Administration (FDA) approval for severe hypertriglyceridemia, we may seek approval for use of MAT9001 in additional indications, including the treatment of patients with mixed dyslipidemia who are already undergoing treatment with a statin, a commonly used class of cholesterol-lowering medications.
Consistent with our strategy first put in place when we filed the investigational new drug (IND) application for MAT9001 in 2014, we intend to pursue a 505(b)(2) regulatory pathway towards NDA approval in the United States. Pursuant to this streamlined development approach, we are permitted to rely, at least in part, on FDA findings of safety and/or effectiveness for a previously approved drug. Based upon written feedback received from the FDA in 2014, we believe this approach will create the opportunity for us to leverage existing data developed with certain existing omega-3 fatty acids to create a streamlined approach to potential approval for MAT9001 for the treatment of severe hypertriglyceridemia (≥500 mg/dL). Simultaneously with those preclinical and clinical studies necessary for approval of this initial indication, we intend to conduct two additional studies designed to highlight the differentiated profile of MAT9001 vs. market leading omega-3 fatty acids and potentially yielding superior data in similar patient populations to those data generated by already-approved omega-3 fatty acids. We believe this dual development strategy will best position MAT9001 to secure an approval to treat severe hypertriglyceridemia as quickly as possible while also positioning MAT9001 as the best-in-class prescription omega-3 therapy as this market and regulatory requirements evolve.
While we advance MAT9001 toward pivotal trials in the cardiovascular space, we are also determined to maximize the value associated with our unique and potentially disruptive lipid nano-crystal (LNC) platform delivery technology. Our proprietary LNC platform delivery technology, licensed from Rutgers University on an exclusive worldwide basis, nano-encapsulates molecules and is designed to render these molecules orally bioavailable, well-tolerated and safe via fusogenic intracellular delivery. We believe the ability of our drug delivery technology to efficiently deliver drugs intracellularly may result in the targeted and safe delivery of pharmaceuticals directly to the site of infection or inflammation as well as the potential to treat a variety of cell-based pathogens, diseases and conditions. We believe our cochleate technology provides us with a highly stable, efficient and broadly applicable drug delivery platform, that has the potential to deliver a broad range of therapies, including small molecules, vaccines, peptides and proteins, as well as nucleic acid polymers in the gene therapy space (e.g., siRNA, mRNA, and CRISPR/Cas-9) in diseases and conditions exhibited by inflammation (e.g., CNS and infectious diseases) as well as intracellular diseases (e.g., intracellular pathogen-related, genetic disorders, and cancer).
Our lead drug candidate based on the LNC platform is MAT2203, an oral formulation of amphotericin B, a well-known and highly-effective, antifungal drug (though traditionally highly-toxic and currently only available in an intravenous formulation) currently used and approved used to treat a variety of invasive, and potentially deadly, fungal infections. MAT2203 has been developed to date with the assistance and financial support of the National Institutes of Allergy and Infectious Disease (NIAID) of the National Institutes of Health (NIH). MAT2203 has been designated as a Qualified Infectious Disease Product (QIDP) with Fast Track Status for the treatment of invasive candidiasis, the treatment of aspergillosis and the prevention of invasive fungal infections in patients who are on immunosuppressive therapy. We have completed two Phase 2 studies of MAT2203 since 2015 and, leading up to and following a meeting with the Office of Antimicrobial Products (OAP) in January 2018, we had been positioning MAT2203 for an initial indication for the prophylaxis, or prevention, of invasive infections in patients who are suffering from acute lymphoblastic leukemia (ALL) who are rendered immunosuppressed due to the therapies being utilized to treat these patients’ leukemia.
While we continue to believe that MAT2203 could become an important solution to the significant unmet medical need to prevent invasive fungal infections in immunosuppressed patients, we believe there are opportunities for a potentially more rapid approval of MAT2203 for the treatment of certain invasive fungal infections in areas of high unmet medical need which can be substantially supported by non-dilutive government funds. In partnership with the NIH, we have conducted numerous preclinical studies of MAT2203 for the treatment of cryptococcal meningitis. In such studies, we observed the potential for MAT2203, utilizing our LNC platform delivery technology, to (a) cross the blood-brain barrier, (b) treat this infection and (c) eliminate the toxicity normally associated with liposomal delivery of amphotericin B intravenously.
This data attracted the attention of several organizations, including the NIH, interested in finding a better treatment option for cryptococcal meningitis. The NIH recently approved and fully funded a grant submission from the University of Minnesota to conduct a clinical study of MAT2203 on patients with cryptococcal meningitis located in Uganda, Africa where this disease is very prevalent among the HIV-positive community. In consultation with the NIH and the University of Minnesota we are finalizing a protocol to commence this study during 2019. We believe that this clinical trial could become a registration-quality trial for an indication for MAT2203 to treat cryptococcal meningitis. We intend to finalize this protocol in the near term and then engage with the FDA to outline a potential pathway for approval of MAT2203 based upon this trial. Given the significant challenge associated with treating cryptococcal meningitis, we believe this indication could position MAT2203 with physicians to become the antifungal of choice for the treatment of all invasive fungal infections given the broad-spectrum nature of amphotericin B. Combined with the oral and targeted delivery and safety profile we believe the LNC platform delivery affords, we believe MAT2203 is well positioned to become a best-in-class antifungal drug. In addition, we believe that a demonstration that MAT2203 can effectively cross the blood-brain barrier in humans in this study could potentially position our LNC platform delivery technology to be utilized with molecules designed to treat diseases of the central nervous system exhibited by inflammation. Developing MAT2203 utilizing non-dilutive, government-sponsored, financing allows us to focus our internal cash resources on MAT9001 while advancing MAT2203 and our LNC platform technology into areas of significant unmet medical need and innovative medicine.
| 2 |
We have been engaged in discussions with various large, well-established and well-financed biotech and global pharmaceutical companies on potential applications of our LNC platform technology in the gene therapy space. Though early stage, these discussions have been based on existing data utilizing our LNC platform technology with complex nucleic acid polymers such as antisense oligonucleotides, mRNA, siRNA, and DNA plasmids. Our success in nano-encapsulating larger nucleic acids such as DNA plasmids, which can be as large as 11 kilobases in length, has also pushed us toward discussions with the NIH and others about utilizing our LNC platform delivery technology in the CRISPR-Cas9 space.
In July 2018, we announced a research collaboration with the National Institute of Neurological Disorders and Stroke (NINDS), part of the National Institutes of Health (NIH), focused on the development of a novel therapy for the treatment of human immunodeficiency virus (HIV) combining targeted antisense oligonucleotides (ASO) and Matinas’ LNC delivery technology. In January 2019 we announced a research evaluation with a top global pharmaceutical company in which our LNC platform technology would be explored in delivering certain nucleic acid polymers, and we continue to pursue additional strategic collaborations with other interested biotech and pharmaceutical partners.
We believe these early stage, proof-of-concept evaluations could provide a more efficient, less expensive pathway to create numerous strategic verticals in areas of innovative medicine relying upon the development expertise and financial resources of well-established pharmaceutical and biotech companies. Our belief is that data from these evaluations could position us to become a licensor of our LNC platform delivery technology to numerous strategic partners better positioned to absorb the risks and costs of drug development while allowing our company to become a royalty aggregator with the potential to generate upfront license, milestone and royalty payments in order to utilize our LNC delivery platform technology.
Strategy
We are focused on creating value through the streamlined and strategic development of MAT9001 for the treatment of cardiovascular and metabolic conditions and the application of our LNC platform delivery technology to solve complex challenges relating to the delivery of small molecules, gene therapies, proteins/peptides, and vaccines. Key elements of our strategy include:
| ● | Strategically advancing MAT9001 into clinical development toward an initial indication for the treatment of severe hypertriglyceridemia (≥500 mg/dL) (SHTG) with the goal of creating additional data further demonstrating the differentiation of MAT9001 from other prescription omega-3 drugs being used to treat a mixed dyslipidemic patient population in a rapidly emerging and expanding omega-3 market. | |
| ● | Expanding application of our lipid nano-crystal (LNC) delivery platform into the gene therapy space through collaborations with sophisticated and well-resourced biotech and pharmaceutical companies in innovative areas of medicine. | |
| ● | Driving MAT2203 to efficacy data in the treatment of cryptococcal meningitis, an area of significant unmet medical need, with the non-dilutive financial support of the NIH. |
| 3 |
MAT9001
Our lead cardiovascular product candidate, MAT9001, is a proprietary prescription-only omega-3 fatty acid composition, comprised of a complex mixture of omega-3 fatty acids, including eicosapentaenoic acid, or EPA, docosapentaenoic acid, or DPA, several other omega-3 fatty acids, and relatively nominal amounts of docosahexaenoic acid, or DHA, and non-omega-3 fatty acids. We believe that based upon MAT9001’s unique composition, which includes more DPA than other known omega-3 fatty acids, it will prove to be differentiated from other existing therapies for the treatment of very high triglycerides, or severe hypertriglyceridemia, and dyslipidemia. Triglycerides are fats that are carried in the blood, together with cholesterol, within lipoproteins. High levels of triglyceride rich lipoproteins are associated with an increased risk of atherosclerotic cardiovascular disease and in the case of severe hypertriglyceridemia, acute pancreatitis. High levels of triglycerides are due to both genetic and environmental factors and are associated with comorbid conditions such as diabetes, chronic renal failure and nephrotic syndrome. Unlike the current approved therapies in this product category, many of which have been repurposed following clinical failures in their originally intended indications, we have specifically designed and developed MAT9001 to treat severe hypertriglyceridemia and dyslipidemia. We believe that the results of these targeted development activities and related clinical investigations may yield an improved therapeutic profile compared to the currently-existing therapies, characterized most importantly by MAT9001’s differentiating mechanistic features associated with its unique high DPA composition and enhanced potency as observed in our head to head clinical trial versus Vescepa.
We are primarily focused on developing MAT9001 through approval by the FDA, with an initial indication for the treatment of severe hypertriglyceridemia. Severe hypertriglyceridemia refers to a condition in which patients have high blood levels of triglycerides (TG ≥ 500 mg/dl) and is recognized as an independent risk factor for pancreatitis and cardiovascular disease. If we receive FDA approval for severe hypertriglyceridemia, we may seek approval for use of MAT9001 in additional indications, including the treatment of patients with mixed dyslipidemia who are already undergoing treatment with a statin, a commonly used class of cholesterol lowering medications. Mixed dyslipidemia refers to a condition in which patients have a combination of both elevated triglycerides (≥200mg/dl), and elevated cholesterol levels. According to the NCEP Guidelines, we estimate that approximately 30 to 35 million Americans have mixed dyslipidemia.
Hypertriglyceridemia and Cardiovascular Disease Market Overview
Hypertriglyceridemia refers to a condition in which patients have levels of triglycerides in their blood above 200 mg/dL. Severe hypertriglyceridemia refers to a condition involving levels of triglycerides equal or above 500 mg/dL. Triglycerides (TG) are fats that are carried in the blood, together with cholesterol and lipoproteins. High levels of triglyceride-rich lipoproteins are associated with an increased risk of atherosclerotic cardiovascular disease. Hypertriglyceridemia is due to both genetic and environmental factors. Environmental factors include obesity, sedentary lifestyle, and high caloric diets. Hypertriglyceridemia is also associated with comorbid conditions such as diabetes, chronic renal failure, and nephrotic syndrome.
The prevalence of hypertriglyceridemia is rapidly increasing in the United States and throughout the world, correlating with the increasing incidence of obesity. Severe hypertriglyceridemia is also associated with markedly increased risk for cardiovascular disease and recent studies have demonstrated that elevated triglyceride levels can be regarded as an independent risk factor for cardiovascular disease-related events such as myocardial infarction, ischemic heart disease, and ischemic stroke.
Heart attacks, strokes and other cardiovascular events represent the leading cause of death and disability among men and women in western societies. More than 1 out of every 3 adults in the United States (approximately 92 million) currently lives with one or more types of cardiovascular disease; an estimated 800,000 new or recurrent coronary events and 795,000 new or recurrent strokes occur each year; an estimated 29 million adults ≥20 years of age have high total serum cholesterol levels (≥240 mg/dL), and an estimated 71 million adults ≥20 years of age have borderline high or high low-density lipoprotein (“bad”) cholesterol, or LDL-C, levels (≥130 mg/dL).
| 4 |
In addition to cholesterol, lipoproteins such as LDL also carry fats in the form of triglycerides. Hypertriglyceridemia refers to a condition in which patients have high levels of triglycerides in the bloodstream and has been reported to be an independent risk factor for cardiovascular disease. Triglyceride levels provide important information as a marker associated with the risk for heart disease and stroke.
Guidelines for the management of very high triglyceride levels (≥500 mg/dL) suggest that reducing triglyceride levels is the primary treatment goal in these patients to reduce the risk of acute pancreatitis. Treating LDL-C remains an important secondary goal. Other important parameters to consider in patients with very high triglycerides include levels of apolipoprotein B (apo B), non-HDL-C, and very low-density lipoprotein cholesterol (VLDL-C).
It is estimated that over 25 million adults in the United States have elevated triglyceride levels ≥200 mg/dL and that more than 50 million adults in the United States have elevated triglyceride levels ≥150 mg/dL. Additionally, approximately 4 million adults in the United States have very high triglyceride levels (≥500 mg/dL).
Mixed dyslipidemia refers to a condition in which patients have a combination of two or more lipid abnormalities including elevated triglycerides, low HDL-C, and/or elevated LDL-C. Both hypertriglyceridemia and mixed dyslipidemia are components of a range of lipid disorders collectively referred to as dyslipidemia. Dyslipidemia has been linked to atherosclerosis, commonly referred to as hardening of the arteries.
Limitations of Current Therapies
Hypertriglyceridemia (HTG) is a prevalent lipid disorder in approximately 25% of the U.S. adult population. Both epidemiological and genetic data have shown associations between HTG and coronary heart disease. Many of those patients are taking statin therapy directed at lowering the risk of cardiovascular disease (CVD) by lowering their LDL-C levels, primarily. Recently, real world administrative database analyses have reported an increased CVD risk as well as direct healthcare costs associated with HTG despite statin therapy and controlled LDL-C compared to those with TG<150 mg/dL.
There is currently no approved prescription omega-3 to lower TG levels in statin-treated patients with mixed dyslipidemia and persistent high (≥200 mg/dL and <500 mg/dL) TG levels due to uncertainty raised by FDA in 2013 regarding the benefit, if any, of drug-induced changes in lipid/lipoprotein parameters beyond statin-lowered LDL-C on cardiovascular risk among statin-treated patients with residually high TG.
Additionally, recent cardiovascular (CV) outcomes trials and meta-analyses with low dose omega-3 fatty acid mixtures containing DHA have not shown substantial benefit in patients receiving contemporary medical therapy, including statins. Due to these failed low dose omega-3 CV outcomes trials, the European regulatory authorities have concluded that omega-3 fatty acid medicines at a dose of 1-gram per day are not effective in preventing further events for patients who have had a heart attack.
TG levels provide important information as a marker associate with the risk for heart disease and stroke, especially when an individual also has low levels of high-density lipoprotein cholesterol (HDL-C) and elevated levels of low-density lipoprotein cholesterol (LDL-C). Multiple epidemiological, clinical, and genetic studies suggest that patients with elevated TG levels (>=200 mg/dL) are at a greater risk of coronary artery disease (CAD) and pancreatitis, a life-threatening condition, as compared to those with normal TG levels. The genes regulating TGs and LDL-C are equally strong predictors of CAD, unlike HDL-C which is not. Other studies suggest that managing and lowering TG levels may reduce these risks. <
| 5 |
Currently Available Treatment Options and Market Opportunity
The dramatic rise in obesity over the last few decades has led to a concomitant increase in cholesterol and triglyceride levels among the population. The collective term for high blood lipid levels such as high cholesterol and high triglyceride levels often used is “dyslipidemia.” Observational studies have resulted in an increased awareness of the critical role that high cholesterol and high triglyceride levels have as a predictor of cardiovascular events. Accordingly, the introduction of new drugs and novel mechanisms of action to lower the risk of cardiovascular events has become a priority. The initial treatment recommendation for patients with dyslipidemia is typically a low-fat diet. If that is not effective, dyslipidemia is then often treated with statins, which account for approximately 80% of all dyslipidemia prescriptions. Statins became a highly successful class of medications for the treatment of dyslipidemia due to their ability to reduce cardiovascular risk in patients at high risk for heart attacks, strokes, and other adverse cardiovascular events. Because of these outcome benefits, the statin utilization rate as compared to the incidence and prevalence of dyslipidemia in the general population, which we refer to as the epidemiology, has risen to almost 40% in the United States. However, the primary activity of statins is in the reduction of LDL-cholesterol levels and they have only modest effects on triglyceride levels. Recognizing that statins alone are not very effective triglyceride lowering drugs, the National Cholesterol Education Program panel recommends the use of more focused therapies to lower triglyceride levels in patients with severe hypertriglyceridemia. Fibrates (a class of amphipathic carboxylic acids), omega-3 fatty acid-based medications and niacin have all been utilized to lower triglycerides levels. In patients with severe hypertriglyceridemia, first-line drug therapy is often a prescription omega-3 or fibrate. Prescription omega-3 based products have been shown to reduce triglyceride levels in the range of 20%-45%.
The treatment rate of hypertriglyceridemia has remained relatively low – below ten percent - compared to the adult population with hypertriglyceridemia. Historically, fibrates such as gemfibrozil (Lopid) and fenofibrate (Tricor or Trilipix) have led the class of treatments of hypertriglyceridemia. However, due to their inability to establish clinical outcome benefits and their limited compatibility with statin therapy, the fibrate utilization rate has remained relatively low and is currently declining. Other products used to treat severe hypertriglyceridemia incorporating niacin as the active pharmaceutical ingredient have not been able to establish additional outcome benefits as compared to statin treatment alone, and are also encountering declining utilization. Because of their lack of outcome benefits, fibrate and niacin use has been mostly concentrated in severe hypertriglyceridemia.
Many omega-3 fatty acid based products have anti-thrombotic and anti-inflammatory effects that suggest effectiveness in inhibiting atherosclerosis in animal models as well as reducing the rate of adverse cardiovascular events in humans at high risk for such events as demonstrated in the JELIS Trial and the GISSI Prevenzione trial in Italy. Furthermore, omega-3 fatty acid based products, either concentrates of both EPA and DHA or EPA alone, have been demonstrated in multiple clinical trials to lower serum concentrations in patients with hypertriglyceridemia. In a recent third-party study, increased levels of EPA and DHA in red blood cells directly correlated with significant reductions in cardiovascular health risks. However, omega-3 fatty acid based medications with significant levels of DHA have been shown to increase LDL-cholesterol levels, which is a negative side effect.
The global prescription omega-3 market has been growing steadily over the last two decades and we estimate the market currently is approaching $2 billion in global sales. The leading omega-3 prescription pharmaceutical products currently approved for the treatment of hypertriglyceridemia are Glaxo Smith Kline’s Lovaza (omega-3-acid ethyl esters, an omega-3 mixture containing mostly EPA and DHA, branded as Omacor in the rest of the world), Omacor and Seacor, very similar to Lovaza and marketed in Europe; and Mochida Pharmaceutical Co., Ltd’s (“Mochida”) Epadel (98% ethyl eicosapentaenoate), the leading Japanese omega-3 product. Recently, a new omega-3 based medication, Amarin’s Vascepa (97% ethyl eicosapentaenoate), was approved and launched in the United States. In addition, Astra Zeneca has an FDA-approved product, Epanova, which has not yet been launched.
| 6 |
MAT9001 Differentiation Strategy
In contrast to certain other omega-3 based prescription products, MAT9001 is not a product repurposed from a previous development program for another disease or condition, as it was specifically designed for the treatment of severe hypertriglyceridemia and mixed dyslipidemia. Specifically, we are pursuing two avenues of differentiation from existing products, including Vascepa and Lovaza:
| 1. | MAT9001 has unique mechanistic features due to its proprietary composition of omega-3 fatty acids, including DPA, which we believe is a key differentiating omega-3 fatty acid component ( i.e. , a component that is neither EPA nor DHA); and | |
| 2. | MAT9001 is designed to have a highly concentrated potency versus other omega-3 products due to its free fatty acid formulation and potentially improved bioavailability relative to other omega-3 fatty acid pharmaceutical products, as demonstrated in our head to head study against Vascepa. Unlike ethyl-ester omega-3 fatty acid formulations, MAT9001 does not require enzymatic breakdown in the small intestine before it can be adequately absorbed. These enzymes are secreted in the intestine in response to dietary fats. Therefore, ethyl-ester omega-3 fatty acids are not optimally absorbed unless they are taken with a high-fat meal, which is contraindicated in patients with hypertriglyceridemia. Because MAT9001 is less reliant on meal-fat content for optimal absorption, it has significantly greater bioavailability than the ethyl-ester form under the recommended low-fat diet conditions. |
We believe that based upon both publicly available preclinical and human data generated with MAT9001, as well as independent data associated with one of the key omega-3 components contained in MAT9001, our product has the potential to:
| 1. | Better control cholesterol, and may decrease low-density lipoproteins, or LDL, cholesterol levels; | |
| 2. | Demonstrate superior reduction across numerous lipid biomarkers, including triglycerides, VLDL, non-HDL and Apo-C3 levels; and | |
| 3. | Produce certain gene regulatory effects, such as the down regulation of HMG-CoA reductase and PCSK9. |
In addition, MAT9001 contains a much lower concentration of DHA than certain competitive omega-3 products, such as Lovaza or Epanova (products with mixtures of mostly EPA and DHA). As described above, these products reduce triglycerides as the main desired effect but also have the negative side effect of increasing LDL-cholesterol levels. This side effect is observed with the use of Lovaza and Epanova in patients with severe hypertriglyceredemia as well as in patients with mixed dyslipidemia. In contrast, products with very low concentrations of DHA, such as Vascepa, have not shown the increase in LDL-cholesterol levels relative to placebo in either the severe hypertriglyceridemia or mixed dyslipedemia patient populations. Omega-3 products containing low DHA levels have also demonstrated reductions in LDL-cholesterol and non-HDL-cholesterol levels. We believe MAT9001’s unique composition will produce differentiating results in reducing both cholesterol and triglyceride levels. Further, based on our product design, we believe that MAT9001 is well-positioned to become a leading treatment for severe hypertriglyceridemia if approved by the FDA.
MAT9001’s free fatty acid form of omega-3 differentiates it from competitors and we believe this distinction leads to numerous clinical advantages. In a head to head study vs. Vascepa, as detailed further below, improved absorption characteristics and bioavailability were observed for MAT9001 as compared to Vascepa. Our PK/PD head-to-head trial compared the bioavailability of MAT9001 and Vascepa and it was observed that MAT9001’s free fatty acid form was less reliant on meal-fat content for optimal absorption than Vascepa’s ethyl-ester omega-3 form, which requires a high-fat meal for optimal absorption. It was also observed that patients on a low-fat diet exhibited five times higher blood plasma levels of EPA relative to Vascepa. Additional benefits of MAT9001’s improved bioavailability may include once-a-day dosing, reduced pill burden and accompanying heightened patient compliance. All adverse events (AEs) reported in this study (whether related or unrelated to study drugs) were mild or moderate in severity. The most commonly reported AEs judged as possibly related to the study drug were dry skin and rhinorrhea. No serious adverse events (SAEs) were reported during the conduct of this study. The study medications were well-tolerated by patients in this study as well.
| 7 |
We believe that MAT9001, with its unique ratios of omega-3 free fatty acids, increased plasma concentrations of EPA compared to Vascepa, potential once-a-day dosing convenience, and observed degree of bioavailability and potential to reduce triglyceride levels as observed in our studies to date, is well-positioned to address significant unmet medical need and become a standard of care in the treatment of hypertriglyceridemia. Furthermore, we believe that MAT9001, due to the gene regulatory effects of DPA, in combination with statins, if approved by FDA, could become a standard of care in patients with mixed dyslipidemia with a prescribing and commercial advantage favoring products with a once per day dosing convenience similar to statins.
Development History
We believe we have optimized the manufacturing process for the active pharmaceutical ingredient of MAT9001 and have completed various preclinical studies and one human clinical trial with the MAT9001 active ingredient. We completed the first preclinical studies of MAT9001 in 2013 with others completed during 2014. In 2015, we announced results from our Head-to-Head PK/PD Trial against Vascepa in which we observed statistical superiority in reducing serum triglycerides, total- and non-HDL-cholesterol, apolipoprotein CIII and PCSK9 levels. The study was a pharmacokinetic and pharmacodynamic, open-label crossover study designed to compare the bioavailability and effects of MAT9001 versus Vascepa on serum triglyceride levels. Forty-two patients were treated with 4 grams/day of MAT9001 or Vacsepa for 14 days, followed by a wash-out period and crossed over to the other treatment arm. Study subjects had fasting TG levels of 200-400 mg/dl without lipid altering therapy, or fasting TG levels of 200 to 350 mg/dL if they were on stable-dose statin monotherapy. MAT9001 was observed to significantly reduce PCSK9 in patients. In this trial, MAT9001 achieved greater median percentage reduction in four of six lipid measures, including total cholesterol, when compared to Vascepa:
| ● | MAT9001 significantly reduced median TG levels by 33.2 percent compared to 10.5 percent for Vascepa (p-value <0.001); | |
| ● | MAT9001 significantly reduced median very low-density lipoprotein cholesterol (VLDL-C) levels by 32.5 percent compared to 8.1 percent for Vascepa (p-value <0.001); | |
| ● | MAT9001 significantly reduced median non-HDL-C levels by 8.8 percent compared to 4.6 percent for Vascepa (p-value=0.027); | |
| ● | MAT9001reduced median HDL-C levels by 11.3 percent compared to 11.1 percent for Vascepa (p-value= 0.337) | |
| ● | MAT9001 reduced median LDL-C levels by 2.4 percent compared to 4.3 percent for Vascepa (p-value=0.116) | |
| ● | MAT9001 significantly reduced median total cholesterol levels by 9 percent compared to 6.2 percent for Vascepa (p-value=0.013). |
MAT9001 also outperformed Vascepa in reductions in apolipoproteins (apo) and PCSK9 as compared to baseline:
| ● | MAT9001 reduced median apolipoprotein B levels by 3.8 percent compared to 0.7 percent for Vascepa (p-value=0.058); | |
| ● | MAT9001 significantly reduced median apolipoprotein AI levels by 15.3 percent compared to 10.2 percent for Vascepa (p-value=0.003); | |
| ● | MAT9001 significantly reduced median apolipoprotein CIII levels by 25.5 percent compared to 5 percent for Vascepa (p-value=0.006); | |
| ● | MAT9001 significantly reduced median PCSK9 levels by 12.3 percent compared to an 8.8 percent increase in PCSK9 levels for Vascepa (p-value <0.001). |
Pre-treatment median values for lipids, triglycerides, apolipoproteins, and PCSK9 levels were measured. Patients were randomized and put on MAT9001 or Vascepa for 14 days. Following the initial treatment period, there was a 5-week washout period, following which patients were put on the other therapy for 14 days. Forty patients completed the trial. MAT9001 met its primary PK endpoint for bioavailability of omega-3 for MAT9001 relative to Vascepa in this study. Statistical analysis demonstrated superiority of MAT9001 over Vascepa for omega-3 bioavailability (baseline adjusted AUC and Cmax, approximately 6-fold higher with MAT9001 on Day 14, with very high statistical significance).
| 8 |
MAT9001 Development Plan
Following announcement of our head to head study vs. Vascepa in 2015, due primarily to cardiovascular regulatory and commercial market conditions, as well as limited financial resources, we determined to slow down development of MAT9001 until such time as data became available from Amarin’s cardiovascular outcomes trial, REDUCE-IT™.
Following the release of data from the Amarin REDUCE-IT trial, we have re-initiated our development efforts and activities for MAT9001. With the support of a world-class team of key opinion leaders, clinicians and regulatory experts we have designed a development program for MAT9001 designed to (a) potentially streamline our path to approval for MAT9001 in its initial indication to treat severe hypertriglyceridemia and, (b) create additional data both head to head vs. Vascepa and in a mixed dyslipidemic patient population in order to position MAT9001 to become the potential best-in-class omega-3 prescription product for the treatment and prevention of cardiovascular conditions. Our regulatory strategy with FDA will include leveraging a 505(b)(2) registration pathway, consistent with the feedback we received from FDA during 2014. We are in the process of reactivating our IND and expect to complete this activity in the second quarter of 2019.
Pursuant to our development strategy, we intend to advance MAT9001 into a series of clinical trials designed to (a) complete those studies required for approval of an initial indication to treat severe hypertriglyceridemia (≥500mg/dL) and, (b) complete additional trials to demonstrate the differentiation of MAT9001 vs. competitive approved omega-3 products and also create the potential for label enhancement in a mixed dyslipidemic patient population (patients with triglyceride levels 200-499 mg/dL).
During 2019 we intend to initiate and complete the following studies (a) a 28-day comparative bridging toxicology study, and (b)subject to any feedback from FDA, a comparative clinical bioavailability study (36 healthy volunteers) with key endpoints and assessments to include PK parameters (e.g., AUC, Cmax, Tmax, t1/2) for total EPA, DHA and DPA and comparison of PK parameters for MAT9001 after a high fat meal and also fasting vs. fed.
Following completion of these studies, we intend to request an End-of-Phase 2 Meeting with FDA to review the data from the completed studies and to gain a Special Protocol Assessment (SPA) on our pivotal study protocol for the treatment of severe hypertriglyceridemia. During this meeting, we intend to present the design of a Phase 3 registration study in SHTG patients. We anticipate, subject to feedback from FDA, that the study will be a placebo-controlled study with two dose groups of MAT9001: 2 gram and 4 gram/day. We anticipate that approximately 270 patients will be randomized 1:1:1. It is planned that MAT9001 will be dosed either once or twice daily without regard to meals. The primary endpoint of the study is anticipated to be change in TG levels at Week 12.
In addition to the studies required for approval to treat SHTG, we intend to conduct additional trials, including a comparative PK/PD study vs. Vascepa and a second Phase 3 trial of MAT9001 as an add-on to statin therapy in patients with high triglycerides (200-499 mg/dL) at risk for cardiovascular disease. We anticipate that our second head to head study vs. Vascepa will generate topline data during the second half of 2020.
| 9 |
MAT2203 - Our Lead Product Using our LNC Delivery Platform Technology
We have leveraged our platform lipid nano-crystal (LNC) platform delivery technology to develop two clinical-stage products that we believe have the potential to become best-in-class drug. Our lead LNC platform product candidate, MAT2203, is an orally-administered LNC formulation of a broad spectrum anti-fungal drug called amphotericin B. We previously had been planning to conduct a Phase 2/3 adaptive-design study of MAT2203 in patients with Acute Lymphoblastic Leukemia (ALL) for the prevention of invasive fungal infections (IFI) due to immunosuppressive therapy. While we continue to believe that MAT2203 could become an important solution to the significant unmet medical need to prevent invasive fungal infections in immunosuppressed patients, we believe there are opportunities for a potentially more rapid approval of MAT2203 for the treatment of certain invasive fungal infections in areas of high unmet medical need which can be substantially supported by non-dilutive government funds. In partnership with the NIH, we have conducted numerous preclinical studies of MAT2203 for the treatment of cryptococcal meningitis. In such studies, we observed the potential for MAT2203, utilizing our LNC platform delivery technology, to (a) cross the blood-brain barrier, (b) treat this infection and (c) eliminate the toxicity normally associated with liposomal delivery of amphotericin B intravenously.
We now plan to initially develop MAT2203 for the treatment of cryptococcal meningitis, one of the most frequent and opportunistic infections in Human Immuno-Deficiency Virus (HIV) patients. Given the high morbidity associated with cryptococcal meningitis in HIV patients, the clinical unmet need is globally very high with the global burden estimated at 1 million cases annually. We believe MAT2203, if approved, to have the potential to become the drug of choice for physicians in the treatment of other invasive fungal infections. We plan to leverage the 505(b)(2) regulatory pathway for MAT2203, in part relying upon FDA’s findings ofthe efficacy of amphotericin B, and anticipate meeting with the FDA in the first half of 2019 to discuss our development plans for MAT2203. We also plan to seek accelerated approval for this indication. We are in the final planning stages for a Phase 2 clinical trial, fully funded by the NIH and conducted by the University of Minnesota at their clinic in Uganda. We believe that this study may have the potential to become a pivotal study to support approval of MAT2203 for the treatment of cryptococcal meningitis, and we also plan to submit an application for Orphan Designation and QIDP Designation during the first half of 2019.
Our second clinical stage LNC-based product candidate is MAT2501, an orally administered, cochleate formulation of the broad-spectrum aminoglycoside antibiotic amikacin which may be used to treat different types of multidrug-resistant bacteria, including non-tuberculous mycobacterium infections (NTM), as well as various multidrug-resistant gram negative and intracellular bacterial infections. In May 2017, we completed and announced topline results from a Phase 1 single escalating dose clinical trial of MAT2501 in healthy volunteers in which no serious adverse events were reported and where oral administration of MAT2501 at all tested doses yielded blood levels that were well below the safety levels recommended for injected amikacin, supporting further development of MAT2501 for the treatment of NTM infections. We have decided to temporarily halt clinical development of MAT2501, in order to prioritize and accelerate the development of MAT9001 and MAT2203 and explore utilization of our LNC platform delivery technology in the gene therapy space.
MAT2203 - Product Profile
MAT2203 is an orally-administered, LNC formulation of amphotericin B (a broad-spectrum fungicidal agent). Little to no clinical resistance has been reported to date with amphotericin B as compared to the rapidly emerging drug resistance seen in other antifungal therapies. Currently, IV-only administered amphotericin B is the only broad spectrum fungicidal; however, it has significant treatment-limiting side effects, most notably nephrotoxicity. The ability to provide amphotericin B orally using our proprietary and novel oral formulation comprising our LNC platform delivery technology, may offer a new and promising alternative for patients and doctors. In a clinical Phase 1 single-dose, double-blind, dose-escalating, pharmacokinetic study of 48 healthy volunteers, oral MAT2203 was observed to be well tolerated with no serious adverse events reported, and without any observed nephrotoxicity. The most commonly reported AEs were nausea and abdominal pain. None of the AEs were related to abnormal laboratory evaluations. All treatment emergent adverse events (TEAEs) were mild except 1 instance of “upper respiratory tract infection” which was moderate in a subject following 800 mg MAT2203. No AEs led to withdrawal. There were no serious AEs. There was one pregnancy (subsequently determined that the conception date was 1 to 2 days prior to dosing) resulting in elective termination from the study. More recently, in our Phase 2 trial of MAT2203 conducted by the National Institutes of Health, four out of four enrolled patients met their primary efficacy endpoint, three patients continue on treatment of which two have been successfully taking MAT2203 for more than two years as part of a long term safety extension, with no evidence of kidney or other toxicity frequently associated with the use of amphotericin B.
| 10 |
Antifungal Market Opportunity
The overall global antifungal market accounted for $10.7 billion in 2015 with estimated annual worldwide sales of prescription systemic antifungal drugs reaching approximately $4 billion. This includes therapies used as active treatment or prophylaxis (preventative) in the inpatient and outpatient setting, therapies used for the treatment of hospitalized patients and therapies used for the treatment of patients who are being discharged from the hospital. We estimate that, each year, there are over 1.5 million cases of invasive fungal infections caused by various species of Candida, Aspergillus and Cryptococcus, the three most common invasive fungal pathogens, globally. The estimated incidence in the U.S. for these conditions is approximately 46,000 for invasive candidiasis, 6,000 for invasive aspergillosis, and 3,000 for cryptococcal meningitis. The rapid progression of disease and high mortality rates (20% - 50%) associated with documented invasive fungal infections often result in antifungal therapy being administered in suspected (unconfirmed) cases or as a preventative measure in patients at high risk. Also, the increasingly widespread use of immune suppressive drugs as cancer chemotherapy or for organ transplantation or treatment of autoimmune disease has resulted in an increasing population of patients at risk for invasive fungal infections. Furthermore, the limited number of systemic antifungal drug classes, consisting of azoles, echinocandins and polyenes, and their extensive use, has led to increased numbers of infections with drug-resistant strains. The Centers for Disease Control and Prevention (CDC) has listed fluconazole-resistant Candida as a serious threat requiring prompt and sustained action and has also identified a rise in echinocandin resistance, especially among Candida glabrata. In June 2016, the CDC issued an extraordinary alert for healthcare facilities and providers to be on the lookout for patients with Candida auris, a multidrug resistant strain with high mortality (approximately 60%). Almost half of C. auris isolates are multidrug resistant to two or more antifungal classes (large majority resistant to fluconazole, 40% resistant to echinocandins). We believe this underscores the urgent need for new agents with demonstrated activity against resistant strains and that can be administered with significantly less toxicity and the potential to discharge patients earlier to reduce hospital stays and associated costs.
Physicians’ options for the treatment of fungal infections are limited by a lack of innovative therapies. Several factors have contributed to the low rate of antifungal drug development, including a previously challenging regulatory environment that necessitated large and costly clinical trials. As a result of this regulatory environment and other factors, the number of antifungals in development has decreased, while anti-microbial resistance has increased.
Our Solution – MAT2203
Our lead anti-infective product candidate, MAT2203, is an application of our LNC platform delivery technology to a broad spectrum anti-fungal drug called amphotericin B. Amphotericin B is an IV administered drug used as a last resort for treatment of systemic fungal infections resistant to triazoles and echinocandins, including resistant candidiasis, cryptococcal meningoencephalitis, and aspergillosis. To date, there have been little to no reports of clinically observed drug-resistance to amphotericin B, further bolstering the use of this compound as the most likely last resort treatment for fungal infections in the foreseeable future. However, the use of amphotericin B is relatively limited because it is currently only available as an IV-administered product and has documented history of severe toxicity (most notably nephrotoxicity). By utilizing our LNC platform delivery technology to nano-encapsulate amphotericin B, there is now an opportunity for the drug to be administered orally with targeted delivery to infected cells, which we believe may have fewer side effects than the currently available IV-formulations of amphotericin B. Our LNC delivery of amphotericin B changes the bio-distribution, resulting in a higher level of the drug at the site of infection and a lower level of circulating amphotericin B. By reducing the amount of circulating drug, our LNC may reduce overall toxicity. Importantly, drug concentrations will be high only in tissues due to the migratory nature of macrophages to inflammatory regions. Based upon our studies to date, we believe MAT2203 has the potential to offer improved safety and reduced toxicity and, as a result, we believe MAT2203 will be able to offer a categorically different formulation that delivers orally administered amphotericin B, directly to the target cell at the site of infection. In collaboration with the NIH, in multiple studies, we have demonstrated in cryptococcal meningitis mouse models that our LNC-delivered amphotericin b, following oral administration, has the ability to successfully cross the blood brain barrier to the site of infection in mice. This demonstration provides important data indicating that our LNC platform delivery technology could become an important delivery solution for a variety of CNS-based disorders and diseases.
| 11 |
We believe that MAT2203 has the potential to become a best-in-class induction, consolidation, and maintenance therapy for the treatment of cryptococcal meningitis in HIV patients by offering the following key benefits.
| ● | Potential to treat resistant pathogens. We believe that MAT2203 has the potential to prevent and treat fungal infections caused by drug resistant fungi, including those resistant to existing azoles and echinocandins, due to amphotericin B’s fungicidal (i.e. killing the fungi) nature and potency against resistant strains and the potential for our cochleate drug delivery platform to provide higher drug exposure early in the course of therapy. | |
| ● | Enabling an all-oral therapy. Cryptococcal meningitis has become the most common cause of adult meningitis in many parts of Africa, where cryptococcosis now rivals tuberculosis in all-case mortality. While long-term survival has improved with widespread use of antiretroviral therapy in high income countries, early mortality remains high. Early mortality rates are often ~ 70% in routine practice where access to diagnostics or medications is limited or unavailable, intracranial pressure is uncontrolled, or in settings where other barriers to the management of cryptococcal meningitis exist. IV administration of amphotericin B deoxycholate is not often possible in resource-limited settings, even when it is available. | |
| ● | Shorter and less costly hospital stays and lower outpatient costs. By providing physicians and patients with access to an orally available, broad spectrum fungicidal agent in MAT2203, there is the potential to reduce hospital costs, which account for over 70% of the overall treatment cost of invasive fungal infections |
The FDA has granted MAT2203 designations for Qualified Infectious Disease Product, or QIDP, and Fast Track for the treatment of invasive candidiasis and aspergillosis and for the prevention of IFIs in patients on immunosuppressive therapy. We are in the process of applying for a fourth QIDP for the treatment of cryptococcal meningitis. We will also apply for Orphan Drug Designation for MAT2203 for the treatment of cryptococcal meningitis. The FDA may designate a product candidate as an orphan drug if it is intended to treat a rare disease or condition, which is generally defined as having a patient population of fewer than 200,000 individuals in the United States, or a patient population greater than 200,000 in the United States where there is no reasonable expectation that the cost of developing the drug will be recovered from sales in the United States. The orphan drug designation provides eligibility for orphan drug exclusivity in the United States upon FDA approval if a product that has orphan drug designation subsequently receives the first FDA approval for a particular active ingredient for the disease for which it has such designation. For a product that obtains orphan drug designation on the basis of a plausible hypothesis that it is clinically superior to the same drug that is already approved for the same indication, in order to obtain orphan drug exclusivity upon approval, clinical superiority of such product to this same drug that is already approved for the same orphan indication must be demonstrated. Orphan drug exclusivity means that the FDA may not approved any other applications, including an NDA, to market the same drug for the same indication for seven years, except in limited circumstances such as if the FDA finds that the holder of the orphan drug exclusivity has not shown that it can assure the availability of sufficient quantities of the orphan drug to meet the needs of patients with the disease or condition for which the drug was designated. Similarly, the FDA can subsequently approve a drug with the same active moiety for the same condition during the exclusivity period if the FDA concludes that the later drug is clinically superior, meaning the later drug is safer, more effective or makes a major contribution to patient care. Orphan drug designation also entitles a party to financial incentives such as opportunities for grant funding towards clinical trial costs, a waiver from payment of user fees, an exemption from performing clinical studies in pediatric patients unless the FDA requires otherwise by regulation, and tax credits for the cost of the clinical research. The QIDP designation, provided under the Generating Antibiotic Incentives Now Act, or the GAIN Act, offers certain incentives for the development of new antibacterial or antifungal drugs, including eligibility for Fast Track designation, priority review and, if approved by the FDA, eligibility for an additional five years of marketing exclusivity. Fast Track designation enables more frequent interactions with FDA to expedite drug development and review. Fast Track designation does not change the standards for approval and we can provide no assurances that we can maintain Fast Track designation for MAT2203 or that such designation will result in faster regulatory review. The seven-year period of marketing exclusivity provided through orphan designation, if granted, combined with an additional five years of marketing exclusivity provided by the QIDP designation positions MAT2203 with a potential for a total of 12 years of marketing exclusivity to be granted at the time of FDA approval. Our plan is to further secure QIDP/Fast Track/Orphan Designation for the initial development target indication of cryptococcal meningitis.
| 12 |
Development History of MAT2203 and Initial Target Indication
MAT2203 was studied in animal model studies of various fungal infections including invasive candidiasis, aspergillosis and cryptococcal meningitis.
The data from animal studies for MAT2203 indicate a side-effect advantage over other amphotericin B formulations, which we believe is based on two phenomena:
| ● | The lipid-crystal nano-particle is a solid particle that does not significantly “leak” its drug content while circulating. The particle releases its medication pay-load only when inside the target cells, and thus appears that the use of MAT2203 does not result in toxicities normally seen in the kidneys when using current formulations of amphotericin B. | |
| ● | Because of this targeted approach, we have been able to increase the therapeutic window on a mg/kg basis as compared to IV amphotericin B formulations. We have observed equivalent efficacy at lower doses as well as been able to use oral doses of up to 10x the highest tolerable IV dose in animal model studies. |
NIH-Conducted Study
In early 2017, we reported interim data from the NIH-Conducted Phase 2a Clinical Study of Orally-Administered MAT2203 for the Treatment of Chronic Refractory Mucocutaneous Candidiasis. At that time, two out of the two patients with long-standing azole resistant mucocutaneous candidiasis met the primary endpoint of the Phase 2a study, achieving ≥ 50% clinical response with treatment of MAT2203. Patient #01 achieved a 57% reduction in clinical symptoms after 8 weeks on therapy while patient #02 achieved an 85% reduction in such clinical symptoms after 6 weeks of treatment. MAT2203 was well tolerated with majority of adverse events observed being mild in severity and mostly unrelated to study drug. Importantly, for both patients renal and liver function parameters remained well within normal ranges during the core study as well as during the first 6-month extension of this study. In July 2017, the NIH/NIAID institutional review board approved continuation of treatment of patients in the study-extension for an additional 6 months, for total extension of up to one year.
In January 2018, the National Institutes of Health (“NIH”) reported positive data from a third patient enrolled in this study. This third patient, with long-standing azole resistant mucocutaneous candidiasis, met the primary endpoint of the Phase 2a study in achieving ≥ 50% clinical response with treatment of MAT2203. MAT2203 was well tolerated with any adverse events observed being mild in severity and unrelated to study drug. With this third positive response, the study has met its statistical hurdle for success. In June of 2018, the NIH reported that a fourth patient had enrolled in the study and had met the primary endpoint in achieving ≥ 50% clinical response with treatment of MAT2203. All four patients had been enrolled in a long-term study extension and the initial two patients have now shown no signs of kidney or liver toxicity over the approximately twenty-four months of being administered MAT2203. The third patient was required to drop out of the long-term safety portion of the study due to the development of an infection that does not respond to amphotericin B. The fourth patient continues in the long-term safety extension for the study. The clinical response to MAT2203 seen in all three patients continuing on drug has been maintained and/or improved during the extension period in addition to patients reporting meaningful quality-of-life improvements.
VVC Study
In late 2017, we announced the topline data from our Phase 2 study in Vulvovaginal Candidiasis (VVC) using MAT2203. In the context of our overall program for MAT2203 with the aim to develop our lead product initially for the prevention of invasive fungal infections in patients who are immunocompromised due to immunosuppressive therapy, our goal was, in addition to further establishing the safety and tolerability of MAT2203, to demonstrate efficacy of MAT2203 through a mechanism involving systemic absorption in a non-life threatening fungal infection. This study concept was consistent with early human efficacy studies in the development of other anti- fungal therapies. This Phase 2 study was not designed or powered to support an indication for the treatment of VVC and therefore supplant fluconazole as the standard of care. The key data generated from this study included additional safety and tolerability data.
| 13 |
In this VVC study, the primary endpoint of safety was met, and it was demonstrated that oral delivery of encochleated amphotericin B is safe and well tolerated without the renal and hepatic toxicities that can be seen with administration of intravenous amphotericin B. Drug-related treatment emergent adverse events in this study were mostly of mild and gastro-intestinal nature and were seen at a rate of 20%, 18% and 2% respectively for MAT2203 200mg, MAT2203 400mg, and fluconazole. Consistent with the safety observations in the NIH study, in this VVC study no drug-related effects on liver function were observed and kidney function parameters stayed within normal ranges during the entire study for all 91 patients treated with MAT2203 for 5 days.
Development Plan
In February 2019, the NIH approved the funding of a planned clinical trial related to a grant application submitted by The University of Minnesota to study MAT2203 for the treatment of cryptococcal meningitis. The clinical study will be conducted in Uganda and has already received the approval of all necessary regulatory authorities. The protocol is in the process of being finalized and the trial is anticipated to consist of two parts with the following design. The initial portion of the trial will be a Phase 1 Study conducted in HIV patients in Uganda without active neurological infection. Subjects in this initial phase of the study will involve a determination of the maximal tolerated dose to arrive at an optimal dose for the second part of the study. A Data Monitoring Committee (DMC) will review the data from the initial phase prior to commencing the efficacy portion of the trial. Upon review of the data, the DMC will make a recommendation to the Ethics Committee/IRB as to the dose to be used in the efficacy portion of the study in patients with active neurological infection. The efficacy portion of the study has been designed as an open-label trial to evaluate the safety, tolerability, and microbiologic efficacy of oral MAT2203 as part of induction and consolidation treatment of HIV-infected patients with cryptococcal meningitis compared with standard intravenously delivered amphotericin B. Participants in the study will be enrolled in sequential cohorts designed to mitigate the risk associated with these very sick patients. Induction treatment in each cohort will start with IV amphoterticin and flucytosine treatment with oral MAT2203 administered as step-down treatment for the initial two cohorts (with earlier step-down to MAT2203 in each subsequent cohort.) The next two cohorts will test the induction of treatment with our MAT2203 product, with step-down treatment to IV amphotericin. The final cohort of patients will have a MAT2203 induction treatment (plus flucytosine) without IV administered amphotericin b. The primary endpoint for this trial will be the rate of cerebrospinal fluid (CSF) Cryptococcus clearance as measured by serial quantitative CSF fungal cultures. This study is planned to commence in the second half of 2019. The trial design is expected to be finalized during 2019 following a meeting with FDA.
We are currently completing a 90-day rat toxicology study to support dosing with MAT2203 beyond the current 28-day tox coverage. The in-life portion of the study has been completed. There have been no signs of toxicity noted to date.
Our plan is to meet with FDA to review our development plan and study design as we intend to conduct this Phase 2 study in patients with cryptococcal meningitis under a US IND. We additionally will discuss with FDA our plans to leverage a 505(b)(2) Pathway, relying, in part, upon FDA’s findings of safety and efficacy of I.V. amphotericin B.
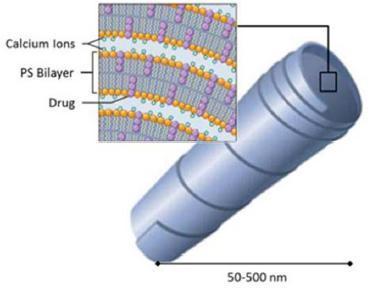
Our Cochleate Platform Delivery Technology
Cochleate lipid-crystal nano-particles are composed of simple, naturally occurring materials: phosphatidylserine (PS) and calcium. They are stable and have a unique multilayered structure consisting of a large, continuous, solid, lipid bilayer sheet rolled up in a spiral or as stacked sheets, with no internal aqueous space (Figure 1). This unique structure provides protection from degradation for “encochleated” molecules. Components within the interior of the cochleate remain intact, even though the outer layers of the cochleate may be exposed to harsh environmental conditions or enzymes.
| 14 |
Figure 1 Cochleate Formulation
The structure is formed when a series of solid lipid sheets engulf drug molecules, a process referred to as “encochleation.” Encochleation, developed by Matinas and Rutgers New Jersey Medical School, involves combining calcium and soy-derived PS, two naturally occurring materials classified as GRAS (generally recognized as safe), through a stirring process to envelop the active pharmacological ingredient. The result is a nano-size encochleated drug formulation (Figure 2).
Figure 2 Formation of Cochleate
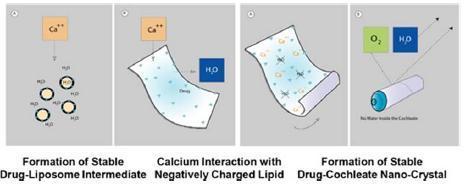
Cochleates have been shown to improve existing drugs by providing 1) cell-targeted delivery; 2) reduced blood levels thereby reducing toxicity; and 3) oral delivery of drugs now only available intravenously. Cochleates work by encapsulating molecules of drugs in a solid, anhydrous, crystalline structure, protecting them as they pass through the gastrointestinal (GI) tract where they cross the mucous membrane. Once the cochleates have crossed the mucosal barrier of the GI tract into the lymphatic system, they are picked up by particle scavenging cells of the mononuclear phagocytic system, such as macrophages and dendritic cells. (Figure 3). Activated macrophages, with drug-cochleate inside, migrate to the site of infection or to the target organ and deliver amphotericin B.
| 15 |
Cells in the mononuclear phagocytic system are immune cells that have the capacity to engulf and destroy numerous potentially pathogenic materials and organisms within the body. These cells are found in almost every site of the body, save a few ‘immune privileged’ sites (e.g. eyes, fetus, and testes). Such cells help with non-specific (innate) immune defenses as well as help initiate specific (adaptive) immune responses, thus they play a critical role bridging the gap between innate and adaptive immune responses. Our core capabilities combine the use of lipids as active pharmaceutical ingredients (API) and the use of lipids in “cochleate-shaped” lipid-crystal nano-particle drug delivery vehicles. Therapeutic applications of our proprietary delivery technology were initially focused on the delivery of several potent and highly efficacious anti-fungal and anti-bacterial agents which are currently still associated with serious side effects, including irreversible toxic effects on kidney and hearing function. We believe our technology has the potential for targeted delivery of these agents, which positions us to be at the forefront of dealing with these very serious problems. We have now also expanded our research and development efforts for our LNC Platform to focus on the delivery of a wide range of therapeutic treatments, in particular those in the oligonucleotide space (siRNA, DNA, antisense DNA, mRNA, and CRISPR-Cas9). We continue to push forward our business development efforts to further expand our collaborations across pharma and biotech companies who have innovative therapies with delivery challenges which may be addressable with our LNC platform delivery technology.
Our LNC technology is currently being used to encapsulate potent anti-infective drugs in tiny lipid-crystals which are selectively picked up by cells in the mononuclear phagocytic system, such as macrophages, and transported to infected cells. These tiny lipid crystals are referred to as “cochleates.” Cochleates have a multilayer crystalline, spiral structure with no internal aqueous space. The structure is formed when a series of solid lipid sheets roll up and engulf drug molecules in between the sheets, a proprietary process referred to as “encochleation”. The result is a lipid-crystal encochleated drug formulation made up of nano-sized particles. We believe our cochleate delivery technology provides an effective delivery mechanism without chemically bonding or otherwise altering the drug. Because the medications are locked in the particles, we believe the exposure to sensitive organs will be reduced, potentially resulting in reduced toxic effects. In summary, we believe this unique technology offers (1) targeted delivery, (2) decreased toxic effects, and (3) oral formulation (even for IV-only medications).
Multi-organ Protection: The key innovation of our cochleate delivery technology is our ability to package medication inside lipid-crystal particles without leaking. Because of their crystal nature, these particles are truly solid and hold on tightly to their medication pay-load. This is where the cochleate delivery technology differs markedly from other lipid-based delivery technology, such as liposomal delivery. Liposomes are liquid delivery systems which typically leak some of their drug content into the circulatory system, thus still exposing vulnerable organs and tissues to potential toxic effects. Keeping potentially organ-toxic medications inside the lipid-crystal particles strongly differentiates our cochleate delivery technology from other drug-delivery approaches.
Targeted Delivery: The size of our individual cochleate lipid-crystals is typically in the range of 50-500 nm. This is very small and by comparison close to the size of a large virus or a small bacteria. Our body produces several cell-types that are designed to remove viruses and bacteria from our system. These cell types, such as macrophages, are part of our immune system and “swallow” the bacteria and viruses they encounter in order to protect us from infections. Because of the size our lipid-crystal cochleate particles and the phospholipid surface structure (the cell membranes of bacteria are also made up from phospholipids), macrophages tend to absorb these cochleate particles very well.
Oral Formulation: Many drugs that are currently on the market are only effective in treating diseases when administered via IV. For example, many anti-infective drugs must be administered via IV in order to be effective. IV administration presents several challenges to care, such as risk of infection, patient discomfort from injections, and higher cost of care than anti-infective drugs that can be taken orally (IV delivery must be performed by a doctor or nurse, often within a very expensive hospital setting). Although several technologies have been used to attempt to convert IV drugs to orally delivered medications, success has been limited due to the difficulty in achieving adequate bioavailability (i.e., the amount of drug that is absorbed into the body) with oral formulation. We believe that the unique cochleate crystal-structure in our platform technology protects the drug from degradation when it passes through the gastrointestinal (GI) tract and that its lipid surface features facilitate the particle to be absorbed into the blood stream. The potential application of our cochleate delivery technology for the delivery of injectable medications offers significant clinical and commercial value if successfully demonstrated in human clinical trials. It is our intent to further validate the LNC Platform technology in our planned cryptococcal meningitis study.
| 16 |
Our cochleate lipid-crystal nano-particle technology changes the delivery of medicines in a unique manner and alters the bio-distribution of these medications by targeting tissues and organs that are affected by infection and inflammation. Besides IV-only anti-infectives such as amphotericin B and amikacin, we have orally delivered in animal studies the influenza vaccine, siRNA, NSAIDs, other anti-infectives such as atovaquone, and many other compounds across multiple therapeutic areas, demonstrating the potential broad application of our technology. We have observed rapid local accumulation in infected tissues, which appear be the result of transport of our drug-loaded cochleates by macrophages and other immune-cells. For example, in a mouse model of invasive candidiasis, comparing orally administered MAT2203 to injected amphotericin B deoxycholate (original drug Fungizone), we observed amphotericin B levels above the minimal inhibitory concentration inside infected organs on day 1 with MAT2203 treatment while such levels were not reached with the injected original amphotericin-deoxycholate product until 3-4 days of treatment. Such kinetics have been seen before with other medications, such as macrolide antibiotics (e.g. azithromycin). It appears from our data that the kinetics of cochleate delivery has similarities to the kinetics of macrolide antibiotics. We expect that additional preclinical and clinical work on the kinetics of our cochleate products will further elucidate the mechanism of cochleate delivery to the site of infection or inflammation.
Strategic Collaborations Using LNC Technology
We believe our LNC platform delivery technology can be used to reformulate a wide variety of molecules and drugs which, (i) require delivery technology to effectively protect molecules and drugs in the body and could benefit from efficient delivery and cellular uptake by target cells, and (ii) are currently only available in IV formulations or, (iii) otherwise experience significant toxicity-related adverse events. Leveraging our cochleate delivery technology, we believe we can develop a robust pipeline of product candidates, either internally or through robust strategic partnerships with pharmaceutical and biotech companies. We have tested a range of pharmaceutical compounds reformulated by our cochleate delivery technology in proof-of-concept animal studies, including oligonucleotides (mRNA, siRNA, DNA plasmids), vaccines, anti-inflammatory agents, NSAIDs and atovaquone. By way of example, in 2016 we received a patent issuance related to LNC compositions directed against expressions of proteins. The allowed patent claims cover our proprietary methods related to the composition and the formation of encochleated siRNA for potential use as therapy for regulating gene expression. We intend to pursue opportunities to develop products, either alone or in partnership with other pharmaceutical or biotech companies, related to this technology and this remains a key part of our strategy to maximize the value of this unique and disruptive lipid-crystal nanoparticle delivery technology.
We continue to actively collaborate with the NIH on a number of therapeutic fronts to further expand the generation of data to support broad use of our LNC platform technology across broad therapeutic treatment modalities. In July 2018, we announced a research collaboration with the National Institute of Neurological Disorders and Stroke (NINDS), part of the National Institutes of Health (NIH) focused on the development of a novel therapy for the treatment of human immunodeficiency virus (HIV) combining targeted antisense oligonucleotides (ASO) and our LNC delivery technology. The goal of this collaboration is to leverage the unique attributes of our LNC technology to safely, effectively and efficiently deliver ASO intracellularly to inhibit Trans-Activator of Transcription (Tat)/viral mRNA translation. Tat is a contributing factor in three major aspects of HIV infection post treatment with antiretroviral therapy (ART): viral replication/latency, chronic inflammation and neurological complications. Tat is a key regulatory protein not specifically targeted by currently available ART. In vitro and in vivo studies will be conducted to determine optimal structures for incorporating ASOs into the LNC technology platform, delivery into target cells and the effective inhibition of Tat and/or viral replication while monitoring Tat-induced cytotoxicity.
In January 2019 we announced a research evaluation with an undisclosed top global pharmaceutical company aimed to evaluate synergistic effects of our lipid-nano-crystal (“LNC”) platform delivery technology with our partner’s nucleic acid polymer technology. Formulations will be developed using our LNC delivery technology which enables the development of a wide range of difficult-to-deliver molecules. Promising formulations will be tested in in vitro and in vivo preclinical studies. For competitive reasons, the agreement stipulates certain confidential provisions, including the pharmaceutical company’s identity, the therapeutic molecule(s), the intended targets and the financial terms of the agreement.
| 17 |
Exclusive License Agreement with Rutgers University
Through our acquisition of Aquarius Biotechnologies Inc., we acquired a license from Rutgers University for the cochleate delivery technology. The Amended and Restated Exclusive License Agreement between Aquarius and Rutgers, The State University of New Jersey (successor in interest to the University of Medicine and Dentistry of New Jersey) provides for, among other things, (1) a license issue fee of $25,000 paid upon execution, (2) an increased equity interest in the company from 5% to 7.5% of Aquarius (prior to our acquisition of Aquarius in the Aquarius Merger), (3) royalties on a tiered basis between low single digits and the mid-single digits of net sales of products using such licensed technology, (4) a one-time sales milestone fee of $100,000 when and if sales of products using the licensed technology reach the specified sales threshold and (5) an annual license fee of initially $10,000, increasing to $50,000 over the term of the license agreement. We also agreed to assume the responsibility to pay required patent prosecution and maintenance fees covering the technology.
Unless otherwise terminated by either party, the term of the license, on a country by country basis, shall be the longer of 7-1/2 years from the date of first commercial sale of a product in a country using the licensed technology or until the expiration of the last-to-expire patent rights licensed under the agreement, whichever is longer. Rutgers has the right to terminate the license agreement if we have not commenced commercial sales of at least one product using the licensed technology within nine years of the effective date of the license agreement.
Intellectual Property
The proprietary nature of, and protection for, our product candidates and our discovery programs, processes and know-how are important to our business. We will seek to protect our products and associated technologies for their manufacturing and development through a combination of patents, trade secrets, proprietary know-how, FDA exclusivity and contractual restrictions on disclosure. Our policy is to pursue, maintain and defend patent rights and to protect the technology, inventions and improvements that are commercially important to the development of our business. Our success will significantly depend on our ability to obtain and maintain patent and other proprietary protection for commercially important technology and inventions and know-how related to our business, defend and enforce our patents, preserve the confidentiality of our trade secrets and operate without infringing the valid and enforceable patents and proprietary rights of third parties. We also rely heavily on know-how and continuing technological innovation to develop and maintain our proprietary position.
Matinas-Owned Intellectual Property Relating to MAT9001
We have sought patent protection in the United States and internationally for our MAT9001 discovery program, and any other inventions to which we have rights, where available and when appropriate. Our policy is to pursue, maintain and defend patent rights, whether developed internally or licensed from third parties, and to protect the technology, inventions and improvements that are commercially important to the development of our business.
Our current patent portfolio relating to MAT9001 and MAT8800 is comprised of two issued patent U.S. patents and one issued foreign patent in Australia. The issued patents claim cover the Company’s proprietary methods relating to triglyceride levels, total cholesterol, VLDL-cholesterol or apolipoprotein C-III by administering a pharmaceutical composition comprising omega-3 fatty acids including eicosapentaenoic acid (EPA) and docosapentaenoic acid (DPA). These patents provide important protection to MAT9001 through 2033. In addition, we have nineteen additional patent applications across four patent families covering the oil composition for MAT9001, other omega-3 fatty acid compositions, as well as formulations of MAT9001 and similar formulations. All of these filed patent applications also comprise methods of use of such oil compositions and formulations. Any patents that may issue from these filed United States patent applications and their counterpart international application covering the MAT9001 drug substance, formulation, and methods for use in treatment would extend protection until at least 2033.
Exclusively Licensed and Matinas-Owned Intellectual Property Relating to Our Proprietary Cochleate Delivery Technology Platform and MAT2203
The patents and patent applications that we exclusively license from Rutgers University provide patent protection for the proprietary chemistry technology used in our process to make our lipid nano-crystal and geodate cochleates and formulate the active pharmaceutical ingredients delivered inside this delivery technology, as in MAT2203, our lead product comprising the LNC platform delivery technology. Pursuant to our license agreement, we acquired rights to a portfolio that currently includes 11 pending applications and 22 issued U.S. and foreign patents, including 15 patents issued within the last 3 years, which extends patent protection until at least 2033. In addition, we have 28 Matinas-owned pending patent applications filed both in the United States and in foreign jurisdictions within the past 3 years. We have chosen to file these patent applications in selected foreign markets that we consider important for our product candidates. These international markets generally include Europe, China, India, Brazil, Russia, Canada, Japan, Korea, Australia and Mexico. These pending patent applications can extend patent protection through 2037. This patent portfolio covers our cochleate delivery system which covers a broad spectrum of technology, including amphotericin B cochleates, geodate cochleates, methods of delivering nutrients or biologically relevant molecules to a host using cochleates, cochleate vaccine compositions and protein-lipid vesicles, small interfering RNA cochleates, mRNA cochleates methods of enhancing the encochleation of hydrophilic molecules and cochleates made with low purity soy phosphatidylserine.
| 18 |
We cannot be sure that patents will be granted with respect to any of our pending patent applications or with respect to any patent applications we may own or license in the future, nor can we be sure that any of our existing patents or any patents we may own or license in the future will be useful in protecting our technology. For this and more comprehensive risks related to our intellectual property, please see “Risk Factors—Risks Relating to Our Intellectual Property and Regulatory Exclusivity.”
In addition to patents, we rely on trade secrets and know-how to develop and maintain our competitive position. For example, significant aspects of our proprietary LNC technology platform as well as the manufacture of certain intermediates utilized in MAT9001, as well as our soft gelatin capsule formulation, are based on unpatented trade secrets and know-how. Trade secrets and know-how can be difficult to protect. We seek to protect our proprietary technology and processes, in part, by confidentiality agreements and invention assignment agreements with our employees, consultants, scientific advisors, contractors and commercial partners. These agreements are designed to protect our proprietary information and, in the case of the invention assignment agreements, to grant us ownership of technologies that are developed through a relationship with a third party. We also seek to preserve the integrity and confidentiality of our data and trade secrets by maintaining physical security of our premises and physical and electronic security of our information technology systems. While we have confidence in these individuals, organizations and systems, agreements or security measures may be breached, and we may not have adequate remedies for any breach. In addition, our trade secrets may otherwise become known or be independently discovered by competitors. To the extent that our contractors use intellectual property owned by others in their work for us, disputes may arise as to the rights in related or resulting know-how and inventions.
We also plan to seek trademark protection in the United States and outside of the United States where available and when appropriate. We intend to use these registered marks in connection with our pharmaceutical research and development as well as our product candidates.
Competition
The biotechnology and pharmaceutical industries are characterized by rapidly advancing technologies, intense competition and a strong emphasis on proprietary products. We face competition from many different sources, including commercial pharmaceutical and biotechnology enterprises, academic institutions, government agencies and private and public research institutions. Many of these companies have far greater human and financial resources and may have product candidates in more advanced stages of development and many will reach the market before our product candidates. Competitors may also develop products that are more effective, safer or less expensive or that have better tolerability or convenience.
MAT9001
Our competitors both in the United States and abroad include large, well-established pharmaceutical and generic companies, specialty and generic pharmaceutical sales and marketing companies, and specialized cardiovascular treatment companies. GlaxoSmithKline plc currently sells Lovaza®, a prescription-only omega-3 fatty acid indicated for patients with severe hypertriglyceridemia, which was approved by FDA in 2004 and has been on the market in the United States since 2005. Multiple generic versions of Lovaza are available in the United States. Other large companies with competitive products include AbbVie, Inc., which currently sells Tricor® and Trilipix® for the treatment of severe hypertriglyceridemia and Niaspan®, which is primarily used to raise high-density lipoprotein cholesterol, or HDL-C, but is also used to lower triglycerides. Multiple generic versions of Tricor, Trilipix and Niaspan are also available in the United States.In 2012, Amarin Corporation received an approval to market its prescription-only omega-3 ethyl ester called Vascepa® for the treatment of severe hypertriglyceridemia
| 19 |
In addition, in May 2014, Epanova® (omega-3-carboxylic acids) capsules, a free fatty acid form of omega-3 (comprised of 55% EPA and 20% DHA), was approved by the FDA for patients with severe hypertriglyceridemia. Epanova was developed by Omthera Pharmaceuticals, Inc., and is now owned by AstraZeneca Pharmaceuticals LP (AstraZeneca). Also, in April 2014, Omtryg, another omega-3-acid fatty acid composition developed by Trygg Pharma AS, received FDA approval for severe hypertriglyceridemia. Neither Epanova nor Omtryg have been commercially launched, but could launch at any time. Each of these competitors, other than Trygg, has greater resources than we do, including financial, product development, marketing, personnel and other resources.
We are also aware of other pharmaceutical companies that are developing products that, if successfully developed, approved and marketed, would compete with MAT9001. Acasti Pharma, or Acasti, a subsidiary of Neptune Technologies & Bioresources Inc., announced in December 2015 that it intends to pursue a regulatory pathway under section 505(b)(2) of the Federal Food, Drug, and Cosmetic Act, or FDCA, for its omega-3 prescription drug candidate, CaPre® (omega-3 phospholipid), derived from krill oil, for the treatment of hypertriglyceridemia.
Akcea Therapeutics/Ionis Pharmaceuticals (formerly Isis Pharmaceuticals), or Akcea/Ionis, in August 2018 announced the receipt of a complete response letter from the FDA for WAYLIVRA™ (volanesorsen), a drug candidate administered through weekly subcutaneous injections, for the treatment of familial chylomicronemia syndrome (FCS). Akcea will continue to work with the FDA on the path forward for Waylivra for the treatment of FCS. Waylivra continues to be developed for the treatment of familial partial lipodystrophy (FPL).
In June 2018, Gemphire Therapeutics announced positive topline results from a Phase 2b trial (INDIGO-1) of its drug candidate, gemcabene, in patients with severe hypertriglyceridemia. Gemcabene is an oral, once-daily pill for a number of hypercholesterolemic populations and severe hypertriglyceridemia.
Zydus Cadila has a Phase 2 development program for its lead molecule, Saroglitazar, in various indications, including severe hypertriglyceridemia in the United States. In August 2018, the Company announced that it had suspended the Phase 2 trial in the severe hypertriglyceridemia indication due to study enrollment issues, while it continues development activities in other indications. The product is approved in India under the name Lipaglyn® for the treatment of hypertriglyceridemia and diabetic dyslipidemia. We are also aware that bezafibrate has been licensed by Intercept Pharmaceuticals to be further developed and potentially launched in the United States market.
MAT2203
Although we believe that our proprietary LNC platform delivery technology, experience and knowledge in our areas of focus provide us with competitive advantages, these potential competitors could reduce our commercial opportunities. For many of our product candidates, we anticipate facing competition from other products that are available on a generic basis and offered at low prices. Many of these generic products have been marketed by third parties for many years and are well accepted by physicians, patients and payers.
We believe that MAT2203 and any other development candidate we may pursue in the future using our proprietary cochleate drug delivery technology platform, paralleled with our scientific and development expertise in the field of drug delivery, provide us with competitive advantages over our peers. However, we face potential competition from various sources, including larger and better-funded pharmaceutical, specialty pharmaceutical, and biotechnology companies, as well as from generic drug manufacturers, academic institutions, governmental agencies and public and private research institutions.
MAT2203 will primarily compete with antifungal classes approved for the treatment of candidemia and mold infections, which include polyenes, azoles and echinocandins. The approved branded therapies for these indications include Cancidas (caspofungin, marketed by Merck & Co.), Eraxis (anidulafungin, marketed by Pfizer, Inc.), Mycamine (micafungin, marketed by Astellas Pharma US, Inc.), Diflucan (fluconazole, marketed by Pfizer, Inc.), Noxafil (posaconazole, marketed by Merck & Co.), Vfend (voriconazole, marketed by Pfizer, Inc.), Sporanox (itraconazole, marketed by Jansen Pharmaceuticals, Inc.), Cresemba (isavuconazole, marketed by Astellas Pharma US, Inc.), Ambisome (liposomal amphothericin B, marketed by Astellas Pharma US, Inc.) Abelcet (lipid complex amphothericin B, marketed by Sigma Tau Phrmaceuticals Inc.) and amphothericin B deoxycholate (marketed by X-Gen Pharmaceuticals, Inc.). There currently are and may be more generic versions of these products available at the time of MAT2203 market approval, which will create added competition. In addition to approved therapies, we expect that MAT2203 may compete with product candidates that we are aware of in clinical development by third parties, such as SCY-078 (being developed by Scynexis, Inc.), CD101 (being developed by Cidara Therapeutics, Inc.) and certain products being developed by Viamet Pharmaceuticals Holdings, LLC, Vical Incorporated and F2G, Ltd.
| 20 |
Manufacturing
We currently contract with one third party manufacturer to supply us with certain of the intermediates used in MAT9001 and a second manufacturer to formulate a third intermediate and supply us with the final drug form. We have a third manufacturer which fills and provides our final MAT9001 capsules. If any of these manufacturers should become unavailable to us for any reason, we have identified a number of potential replacements, although we might incur some delay in qualifying such replacements. We expect to add additional suppliers and manufacturers for both the intermediates and final MAT9001 drug product as we advance MAT9001 further into clinical development.
We currently lease and operate in-house manufacturing capabilities for our lead LNC platform delivery technology product candidate, MAT2203, and for our LNC platform discovery programs in the gene therapy and vaccine spaces. While sufficient to produce the clinical supplies of product necessary to conduct our ongoing clinical trials and potentially early commercialization of MAT2203, we may need to expand our internal manufacturing capabilities in the future. If we are not able to retain our current manufacturing facilities and if we do not develop additional in-house manufacturing capability for our MAT2203 and product candidates sufficient to produce product for commercialization of these products, we will need to develop relationships with third-party manufacturers for the manufacture of our product candidates which could be time consuming and expensive.
There are a number of potential third-party suppliers for amphotericin B, the generic active pharmaceutical ingredients in our lead clinical stage product candidate – MAT2203. Although to date we have not entered into formal supply agreements to secure sufficient supply of amphotericin B to support our clinical programs for MAT2203, we believe we will be able to secure supply of amphotericin B to support our clinical programs for MAT2203 and from one or more third-party suppliers. As we move through development for our product candidate, we expect to enter into long term supply arrangements for key active pharmaceutical ingredients.
Sales and Marketing
We currently do not have any sales and marketing infrastructure. We plan to retain U.S. marketing and sales rights or co-promotion rights for our product candidates for which we receive marketing approvals, particularly in situations where it is possible to access the market through a focused, specialized sales force. For situations in which a large sales force is required to access the market, and with respect to markets outside the United States, we generally plan to commercialize our product candidates through collaborative arrangements with leading pharmaceutical and biotechnology companies.
Implications of Being an Emerging Growth Company
We are an “emerging growth company,” as defined in the Jumpstart Our Business Startups Act of 2012. We will remain an emerging growth company until December 31, 2019, or until the earliest of (i) the last day of the first fiscal year in which our annual gross revenues exceed $1.07 billion, (ii) the date that we become a “large accelerated filer” as defined in Rule 12b-2 under the Exchange Act, which would occur if the market value of our common stock that is held by non-affiliates exceeds $700 million as of the last business day of our most recently completed second fiscal quarter, or (iii) the date on which we have issued more than $1 billion in non-convertible debt during the preceding three year period.
| 21 |
For as long as we remain an “emerging growth company,” we intend to take advantage of certain exemptions from various reporting requirements that are applicable to public companies that are not “emerging growth companies” including, but not limited to, not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act, reduced disclosure obligations regarding executive compensation and financial statements in our periodic reports and proxy statements, and exemptions from the requirements of holding a nonbinding advisory vote to approve executive compensation and shareholder approval of any golden parachute payments not previously approved. We will take advantage of these reporting exemptions until we are no longer an “emerging growth company.”
Corporate Information
We were incorporated in Delaware under the name Matinas BioPharma Holdings, Inc. in May 2013. We have two operating subsidiaries: Matinas BioPharma, Inc., a Delaware corporation, and Matinas BioPharma Nanotechnologies, Inc., a Delaware corporation. Nereus BioPharma LLC, a Delaware limited liability company (and Matinas BioPharma’s predecessor) was formed on August 12, 2011. On February 29, 2012, Nereus BioPharma LLC converted from a limited liability company to a corporation and changed its name to Matinas BioPharma, Inc. In July 2013, Matinas BioPharma, Inc. merged with and into a wholly-owned subsidiary of ours, thereby becoming a wholly owned subsidiary of ours. On January 29, 2015, we acquired Aquarius Biotechnologies Inc. which was subsequently renamed Matinas BioPharma Nanotechnologies, Inc.
Our principal executive offices are located at 1545 Route 206 South, Suite 302, Bedminster, New Jersey 07921, and our telephone number is (908) 443-1860. Our website address is www.matinasbiopharma.com. Our website and the information contained on, or that can be accessed through, our website will not be deemed to be incorporated by reference in, and are not considered part of, this prospectus. You should not rely on our website or any such information in making your decision whether to purchase our securities.
Recent Results
At December 31, 2018, the Company had approximately $13.0 million in cash and cash equivalents, including restricted cash. This amount is preliminary, unaudited and subject to the completion of the audit of the Company’s consolidated financial statements as of and for the year ended December 31, 2018 (Audited 2018 Financial Statements). As a result, this amount may differ from the amount that will be reflected in the Audited 2018 Financial Statements. Additional information and disclosures are required for a more complete understanding of the Company’s financial position and results of operations as of December 31, 2018.
| 22 |